Why do we think Perturb-Map is better than most animal studies?
How do we use Perturb-Map to find the overlap between mouse biology and human biology?
TL;DR: Human data is directly related to what drug development demands, but is difficult to generate and perturb. Mouse data allows us to discover causality and is easy to generate, but is only a proxy for what matters. What has never existed is causal data that speaks in human terms. We’ve built the system that creates it: a combination of wet-lab innovations and machine-learning innovations. The first, Perturb-Map, is a multiplexed in vivo perturbation platform that can test hundreds of genetic knockouts in the same mouse with full spatial resolution. The second is TARIO-2, a foundation model we’ve trained exclusively on human cancer tissue, that can convert H&E to whole-genome spatial transcriptomics. We’ve found that we can apply TARIO-2 directly to H&E derived from Perturb-Map experiments, yielding human-centric tumor microenvironment characterizations from animal data, with no re-training required.
In other words, we can read the results of multiplexed mouse experiments through a lens that only understands human biology.
The combination of Perturb-Map and TARIO-2 is something we call ‘Perturb-MARS’ (Multi-species Alignment and Reasoning on Spatial data), and it allows us to answer not only typical preclinical target discovery questions, but also ones that the current preclinical apparatus is structurally incapable of answering, such as combination therapy exploration; e.g. ‘what is the next PD-1 bispecific target after VEGF and what population is it active in?’. All the while, the readout stays grounded in human-specific genes. We are actively looking for partners here—reach out to [email protected] to learn more.
Every life-saving oncology drug on the market first proved itself in a mouse. And yet, the dominant instinct in the field is to abandon animal models entirely, because 95% of the time, what works in a mouse doesn’t work in a human. Why is this? The standard diagnosis is that mouse biology is a bad model for human biology. This isn’t entirely wrong, but we think the field has it backwards. The problem isn’t the mouse, not entirely. The bigger problem is that we’ve always read mouse experiments with mouse-native readouts—mouse gene expression, mouse protein levels, mouse immune cell counts—and then hoped the translation would sort itself out.
Rather than making the animal more human, what if we changed the lens by which we interpret the animal data?
Last month, we released a post on TARIO-2, an internal foundation model trained exclusively on human cancer tissue. Given an H&E-stained human tumor section, TARIO-2 can predict the spatial gene expression, telling you what genes are expressed across the tissue.
We’ve discovered that TARIO-2 generalizes from human to mouse H&E.
In other words, we’re able to feed it the H&E histology of a mouse tumor, and the model is able to predict what a human tumor with similar morphological features would express at the transcriptomic level. What comes out of the model is not a mouse gene readout, but rather the projection of that mouse experiment into a human biological coordinate system. And when combined with an in vivo genetic perturbation platform called ‘Perturb-Map’, we’ve found that the resulting human-space projections of the mouse H&E are both accurate and have clear use-cases for several clinically relevant tasks.
In this essay, we plan to explain this whole process.
First, how Perturb-Map works. Second, why we believe the method gives far more useful results than most oncology animal studies. Third, and most excitingly, via TARIO-2, we’ve used the method alongside our human tumor foundation models to view mouse data with a human-centric lens; something we’re calling ‘Perturb-MARS’ (Multi-species Alignment and Reasoning on Spatial data). Finally, we’ll end with how this methodology can be used to tighten the currently slow feedback loops between wet-lab and computational predictions, allowing us to train even better generative models of human biology.
The original paper for Perturb-Map was published in Cell in March 2022 by Maxime Dhainaut and colleagues. He was recruited to Noetik shortly after and has directed work on the platform since then, massively scaling it up.
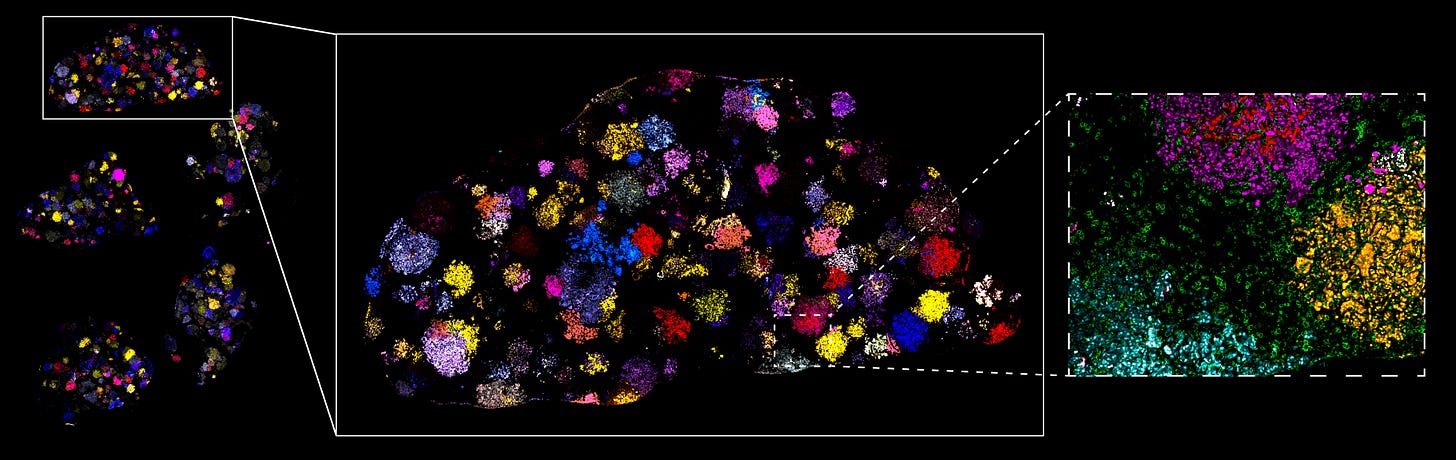
The process starts with cancer cells that are engineered to express unique protein barcodes called “Pro-Codes”. These barcodes consist of triplet combinations of peptide tags, and each unique triplet combination identifies a specific ‘perturbation’, or deliberate genetic change. For instance, a cell expressing tag 1, 2, and 3 together might carry a CRISPR knockout of gene X, while a cell expressing tags 1, 2, and 4 carries a knockout of gene Y. We do this at immense scales, creating hundreds of tumors that each carry their own, distinct mutation; alongside some control cancer cells.
Next, we pool the sum total of these cancer cells together, and inject them into a single mouse in a way that gets them to the tissue of origin from which they were derived (e.g. for lung cancer cells, injection into the tail vein lets them seed in the lung; for breast cancer lines, cells are injected directly into the breast tissue). If this is in a ‘treated’ mouse, we could also administer any arbitrary cancer drug shortly afterwards. After several weeks, the organ—now seeded with vast numbers of genetic mutant cancers—is resected.
Finally, we stain the lung tissue with antibodies that recognize the Pro-Codes, allowing us to directly visualize each genetically perturbed tumor population and trace the growth behavior of every single mutation, using the aforementioned control cancer cells as a baseline. And, if we elected to treat the mouse with an anti-cancer drug, that’d give us additional information as to how the drug affected the tumor microenvironment.
In one fell swoop, this allows us to understand the heterogeneity of each tumor’s response to drugs, assess the relative fitness of different genetic alterations, and potentially identify new cancer targets entirely. All this, at orders-of-magnitude higher scales than typical, single-throughput animal experiments.
Animal data does not have a good reputation. In fact, it has an awful reputation, with many papers’ conclusions often being waved off because ‘it’s based on mouse data’. This is understandable. But we think that its tarnished reputation is sometimes conflating two, completely independent things.
The first one is the inability for mouse physiology to capture human physiology, which, yes, is difficult to solve. We’ll explain more in the next section how we think we’ve put a dent in this problem, but also realize that there are absolute limits in the pharmacologic transferability from one organism to another.
The second is the animal data itself being captured badly, either through flawed experimental design, difficulty in replication, and inability to scale. We think that this second bit is both entirely fixable and accounts for a fair bit of why so many researchers distrust animal data. This section is meant to offer our take on this.
So, why do we think Perturb-Map is better than most animal studies?
First, and biggest, the ability to multiply speed and reduce cost. Traditional animal studies test one, or maybe two, genetic perturbations per cohort of mice. This means that if a researcher wants to understand how 50 different cancer genes affect tumor growth, they’d traditionally need 50+ separate groups of mice. Hundreds of animals, years of work, and enormous costs. With Perturb-Map, all 50 mutations (plus controls) can be tested simultaneously in the same mouse, in the same organ, at the same time. And there is evidence that we can scale this up to hundreds of mutations, allowing us to explore combinatorial mutational space.
Second, and relatedly, this method dramatically attenuates inter-animal variability. In a traditional study, if you want to compare fifty knockouts, you need fifty cohorts of mice, and any differences you observe are entangled with per-animal confounders: vascular supply, microbiome, stress levels, injection technique, and more. With Perturb-Map, all fifty perturbations coexist in the same host, so their relative rankings are controlled by construction. Drug-treatment comparisons still involve a treated and an untreated animal, so some inter-mouse variability re-enters there, but each mouse carries hundreds of knockouts acting as an internal control panel. This makes global per-mouse offsets identifiable and correctable in ways that a single-knockout-per-mouse design cannot match.
Third, this method allows you to retain in vivo spatial resolution. You may realize that Perturb-Map sounds a lot like Perturb-Seq. Perturb-Seq is a related, older method that also allows for multiplexed genetic perturbations. So why aren’t we using that? The problem with Perturb-Seq is that it forces one to dissociate tumor cells to tell which one has which mutation. This is a big problem! A tumor is not a bag of cancer cells floating around. There is an incredibly important, delicate structure to the whole thing. Perturb-Seq, by necessity, destroys all of that. Perturb-Map, in contrast, preserves that spatial information about tumor microenvironments, and the original paper behind it compiled a fairly large list of insights that literally could not have been gained without it.
Fourth, and finally, we place the tumors in their natural environment. There is a somewhat bizarre practice in the oncology field of doing animal studies by injecting tumors just underneath the skin, or what is often referred to as ‘subcutaneous tumors’. This is opposed to ensuring the tumor eventually reaches whatever the tissue of origin is: lungs for lung cancer, pancreas for pancreas cancer, and so on. Using subcutaneous tumors is cheap, fast, and easy to do. It also, unfortunately, yields a cancer microenvironment state that barely resembles that of a tumor grown in its natural environment. We simply don’t do subcutaneous tumors. This, to be fair, is not really related to Perturb-Map; it’s just good experimental hygiene that we enforce across our entire platform.
And that’s why we think Perturb-Map is worth using.
Just because Perturb-Map has good experimental design, reduces variability, is scalable, and retains spatial resolution does not allow it to magically recapitulate human biology. Where are we deriving this understanding of where mouse biology—derived via Perturb-Map—actually maps onto human biology?
A few weeks back, we had a post about TARIO-2, a foundation model trained entirely on human cancer tissue. It can predict whole-plex spatial transcriptomics from H&E-stained human tumor sections.
And when we ran the perturbed mouse tumor H&Es from Perturb-Map through TARIO-2, it worked. The model predicted human gene expression patterns from mouse tumors, despite never having seen a mouse cell during training. Here is an example of it predicting CEACAM6 in mouse H&E.
Importantly, CEACAM6 is a human gene that has no functional ortholog in mice. And you’ll notice it was predicted to be especially high in only small fractions of this H&E slide, which is composed of many genetically different tumors. In other words, the model is likely recognizing the existence of multiple different phenotypes.
Are these phenotypes correct? We’ll discuss that in a bit, but it’s important to first grasp what is actually going on here. TARIO-2 is not translating mouse genes into human genes, nor does it have some emergent understanding of cross-species orthologs. In this specific case, it is instead picking up on a particular morphological mouse phenotype—produced by a genetic knockout—and recognizing it as resembling a human tumor state associated with high CEACAM6. We are, really, treating morphology as a bridge. And morphology may be a surprisingly universal substrate; a shared visual language of tumor states that persist across species even when the underlying genes are not shared.
This combination of Perturb-Map and TARIO-2 is something we’ve termed ‘Perturb-MARS’, or Multi-species Alignment and Reasoning on Spatial data.
But again: how do we know this actually works, and it isn’t just hallucinating plausible-looking predictions? Consider the below t-SNE.
Each dot represents one of the ~600 knockouts in the screen. Its position is computed by running all mouse H&E images containing that knockout through TARIO-2, aggregating the model’s predicted human gene-expression profiles across those tumors, and projecting the result into two dimensions with t-SNE. In this space, nearby dots indicate knockouts whose tumors produced similar inferred human-space microenvironment states.
Our first proof point that this is telling us something meaningful is that the t-SNE is primarily organized by some clinically relevant principle: immune-infiltration. ‘Cold’ tumors, or ones that have low immune infiltration, are consistently on the left side, and hot tumors consistently on the right. If TARIO-2 predictions on mouse H&E were largely random, we would not observe such legible patterns.
Second, and more importantly, some of the knockouts we included in the library have well-established effects on the human immune microenvironment. In yellow, we’ve highlighted these genes. Some of them, when knocked out, should lead to cold tumors (B2M and STK11), or hot tumors (CTNNB1, SOCS1, and PTPN2). And we find that TARIO-2 accurately captures the behavior of these knockouts, correctly placing cold-inducing knockouts on the left, and hot-inducing knockouts on the right.
That these readings align with the literature is, at minimum, compelling validation that the system is capturing real biology rather than generating plausible-looking noise.
And the vast majority of the Perturb-Map screen is composed of tumor knockouts whose effects on the tumor microenvironment have never been characterized spatially! The number of knockouts profiled here, each with full spatial resolution of its impact on the surrounding microenvironment, simply has no precedent. Below, we show an expert-annotated clustering of the TARIO-2 gene predictions, graphed onto the t-SNE, demonstrating that two other tumor environmental niches exist. And likely many more beneath them.
One note before we get to what this is useful for: spatial perturbation profiling is not, strictly speaking, unique to us. What is unique is the readout of this process. Ours is H&E. This is the cheapest, most scalable, and most universally collected assay in all of oncology. Every cancer biopsy ever taken has an H&E slide sitting in a pathology archive somewhere; no spatial transcriptomics platform, ours included, can say the same. The scale of this screen is not enabled by clever sequencing chemistry or bespoke imaging infrastructure, but rather by a stain that costs a few dollars a slide, and a foundation model that does the interpretive work downstream. Moreover, it would be difficult to replicate this work, given that it requires a model trained on thousands of richly characterized human tumor samples.
Moving on: what is the therapeutic application of what we have built here?
One naive way to use this is to simply identify gene knockouts that consistently push tumors toward immunologically hot microenvironments, and nominate them as therapeutic targets. After all, if knocking out GENE-X reliably converts a cold tumor into one teeming with T-cell infiltration, that’s a reasonably strong signal that inhibiting gene X’s protein product could sensitize tumors to immunotherapy. Some of these are already known, e.g., the above genes in yellow, but others aren’t. We are actively exploring novel cancer targets that surfaced as a result of this exploration.
But the far more interesting use of this system is something that falls out naturally from Perturb-Map’s experimental design: because we can run the same panel of genetic knockouts in parallel, once in an untreated animal and once in a drug-treated animal, we can directly compare where each knockout sits in the t-SNE space with and without treatment.
The result, for any given knockout, is a vector—the displacement between its untreated and treated positions—that tells us how the drug reshaped the microenvironment conditional on that specific genetic background. Consider these plots:
Here, each arrow represents the t-SNE displacement between a knockout’s position in an untreated mouse and the same knockout’s position in a treated mouse (anti-PD-1). This is overlaid on a landscape colored by predicted cytotoxic T-cell activity (GZMB expression), which, as it increases, is a rough proxy for ‘the tumor responding to the drug’ (and is functionally extremely similar to the aforementioned immune infiltration signature).
We believe this is extremely useful for patient stratification and combination therapy identification.
For the former, we’re able to ask which genetic backgrounds of a tumor are likely to respond to a given therapy before a patient is ever dosed, not as retrospective enrichment of a trial population, but as a mechanistic basis for deciding in advance whom to enroll and whom not to.
For the latter, we’re able to explore combination therapy candidates: gene targets whose inhibition synergizes with checkpoint blockade to produce an immune response neither achieves alone. For instance: the left plot above parallels the well-established result that VEGFA x PD-1 bispecifics work well, and the redacted GENE-X on the right plot is a known oncology target now being explored in combination with checkpoint blockade.
And interestingly, we can explore not only combination therapy synergy, but also antagonism. As in, identify knockouts that, under treatment, move in the wrong direction. These knockouts actively shift away from immune activation upon PD-1 treatment, moving into transcriptomic neighborhoods associated with immune suppression. This means that for patients whose tumors harbor the corresponding phenotype, the standard-of-care practice of combining PD-1 checkpoint therapy with an inhibitor of that target is potentially worse than doing nothing at all.
Below are two knockouts from this ‘treatment antagonizing’ category.
Interestingly, GENE-B on the right-most plot was identified in a 2024 paper as an emerging therapeutic target for a high-mortality cancer! Our data corroborates the monotherapy case, as the untreated knockout sits in an inflamed tumor microenvironment. But the treatment panel adds something no published work has flagged: combining a drug against this target with standard checkpoint blockade may worsen the patient’s condition.
The utility of being able to stratify patients better is something we’ve written about before, but being able to explore combination regimes at scale is a new capability of ours. How practically useful is this?
Consider daraxonrasib. A few weeks back, Revolution Medicines announced the topline results of their Phase 3 trial for this drug. In previously treated metastatic pancreatic cancer, it nearly doubled median overall survival, from 6.7 months to 13.2 months. That is an extraordinary result in a disease where extraordinary results are rare. But it is still not enough. Most patients still progress, developing resistance to the drug. It feels likely that discovering the ideal drugs to combine with daraxonrasib is a promising path forwards, given early preclinical research on therapies with similar mechanisms. But the unexplored space has and likely will remain enormous: immune targets, stromal targets, and so on. Perturb-MARS would allow us to explore that space at scales and efficiency that no other assay on the planet could.
How will this evolve? What we’ve described is one instantiation of the platform, not how it scales. In this round of Perturb-Map, we screened ~600 total perturbations, ~50 perturbations per animal, ~15 animal replicates per mutation, each one receiving one of four treatments, all on NSCLC subtypes. But there is no theoretical limit to us expanding the number of perturbations per animal, the types of treatments used, or the cancer types we test.
This all said: it’s worth pulling back to what we ultimately care about.
Noetik’s goal is to build a simulator of human cancer biology. Something that, given a tumor and a candidate intervention, can predict what will happen inside the tissue at single-cell resolution. We’ve previously written about one example of pushing such a model in a genuinely generative direction: using OCTO-VC to identify novel targets whose modulation would shift the surrounding T-cells into a more cytotoxic state. This is asking the model to predict what would happen to a tumor under a condition that likely few, if any, patients in its training set were ever observed under.
There is a structural limit to how far this kind of inference can be pushed on human data alone.
Historically, the solution here is post-training on verifiable data. LLMs have been very lucky in that two of the most economically consequential domains they are asked to operate in—code and mathematics—happen to come with cheap, automated verifiers built in. Biology, unfortunately, lacks anything similar.
Perturb-MARS is the closest substitute we know of.
Compared to verifiers in code and math, the throughput is lower, the verification is noisier, and what comes back is a continuous measure of agreement rather than a binary pass/fail. But the structural shape is right. If TARIO-2 predicts that knocking out gene X should drive the surrounding T cells into a particular cytotoxic program, we can take the murine ortholog of that knockout, run it through Perturb-Map, and ask TARIO-2 to read the resulting tumor in human-coordinate space. Once enough predictions and results accumulate, what we have is a signal we can train on. A foundation model post-trained on this kind of feedback should, in expectation, end up more accurate on human biology than one trained on human data alone, because it has been forced to make and revise causal predictions, rather than only ever describing what the world has happened to record.
This is the only end-to-end system we are aware of that connects all three pieces a real simulator in biology needs. Specifically, a model that proposes interventions in human-space biology, a wet lab that runs them in intact living tissue, and a readout that lets us check the prediction in the same coordinate system the model was speaking in. Each round of perturbations expands the universe of predictions we have actually tested, rather than the universe of predictions we have only made, thereby allowing our generative models to reach far beyond what is encapsulated by either the literature or associative snapshots.
How exactly we consume this signal in the long run is something we’re actively researching today. What we feel confident about is the shape of what is being assembled, and the fact that no current analog exists.
Our team is actively seeking partners interested in this work; we already have a rich database of what phenotypes result from different perturbations, and can trivially generate far more. Reach out to [email protected] to start that conversation.
This article would not be possible without significant contributions from Maxime Dhainaut, Michela Meister, Andrew Benz, Keith Mitchell, Lucas Cavalcante, Emily Corse, Ron Alfa, Daniel Bear, Dan Millman, Lacey Padron, and Eshed Margalit.